Thank You for your interest in Prolotherapy, to make an initial consultation
effective and informative please follow the steps below.
* Read all available information about Prolotherapy online, and the information
provided in this packet
* Prepare a list of questions you may have for the doctor.
* Please obtain a copy of any radiological images (XRAY, CT SCAN MRI). You
should be able to request a copy of the radiology imagines from the Radiology
Department.
* Please obtain a copy of your latest blood test results including complete blood
count (CBC) & Basic metabolic panel (BMP)
* Please provide a list of current medical conditions including any allergies and/or
food sensitives.
* Please provide a list of current medications along with the dosage and frequency.
Along with any over the counter medication/supplements you may be taking.
What You Can Expect:
The initial consultation will be focused on taking detailed history and conducting a
musculoskeletal physical exam to assure the appropriateness of Prolotherapy. Your
diagnostic test/s will be reviewed and taken into consideration along with any
medications and/or supplements you are taking. Patients are encouraged to bring a
family member or friend to the consultation to ensure that all possible questions
have been addressed during the consultation.
*PLEASE NOTE: Treatment will not be performed the day of
consultation.
*The cost of the initial consultation is $375.00. Insurance does NOT
COVER PROLOTHERAPY.
Absolute Contraindications for prolotherapy:
Hardware from prior surgical procedure in the affected area
Currently undergoing treatment for cancer
Currently undergoing treatment for autoimmune condition and taking
“immune modulating therapies”
“Full thickness” tendon tears
Any evidence of infection
Pregnancy
FACTS REGARDING PROLOTHERAPY:
Prolotherapy (Regenerative Injection Therapy) was originally used by Hippocrates
over 2500 Years ago to heal a javelin’s shoulder injury. This concept was
investigated, utilized, and refined by Dr. George Hackett, MD over 70 Years ago.
The cause of a great deal of musculoskeletal pain is related to chronic laxity or
relaxation of the connective tissues (ligaments and tendons), which normally
control joint movement and provide support for standing, sitting, and locomotion.
These tissues are moist commonly damaged by trauma and overuse and the initial
healing response may not adequately “weld” the structures back together.
Furthermore, anti-inflammatory medications (Ibuprofen, naproxen) also interfere
with the body’s natural healing process, which may result in chronic weakness/
laxity of these ligaments. These once elastic ligaments and tendons “lose” their
elasticity. Other contributing factors to ligamental laxity are hormonal changes,
such as pregnancy. This is physiologic in preparation for childbirth. However often
times the ligaments and/or tendons do not restore their elastic properties fully.
Other individuals are predisposed because of genetic variations such as Ehlers
Danlos Syndrome or other connective tissue disorders resulting in joint
hypermobility or “double jointedness.”
The result of reduced structural stability is a chronic sprain of the ligament and
tendon fibers which are connected to the extremely sensitive periosteum of the
bone which through nerves sends pain signals to the brain. To compensate for the
instability, the corresponding muscles go into spasm resulting in pain and stiffness.
There lies the paradox: muscle stiffness is the result of joint hypermobility.
Prolotherapy Injections contain dextrose, local anesthetic, and sterile water.
Injections are targeted into the joint space as well as around the joint space (where
ligament/tendon connect to bone – enthesis). The solution is hyperosmolar, causing
a controlled injury and healing response, which results in growth (Proliferation) of
the tissues. In other words, we are injuring the area to stimulate your body to heal.
Over time, the weakened tissues get stronger resulting in increased stability, less
muscle tension/spasms, improved range of motion and eventually less pain.
Numerous studies have shown a success rate of 80-90% over thousands of patients
with success judged as at least 50% reduction in pain.
Typically, injection treatment sessions are administrated 4-6 weeks apart with an
average number of 4-6 injection sessions required per patient. This is very much a
case by case basis. It is important to remember that everybody heals differently and
we will go into your specific situation during the consult. However, the response is
directly related to the speed of healing within our bodies. Generally, young, healthy
people who good nutritional status will heal faster than someone in poor health.
Publications Describing Prolotherapy
1) Hackett, George A., and Henderson, Donald G., “Joint Stabilization: An
experimental, Histologic Study with comments on the Clinical Application in
Ligament Proliferation” American Journal of Surgery, 89 (May 1955),
PP.968-973.
2) Klein, Robert G. Proliferation Injections for Low Back Pain: Histologic
Changes of Injected Ligaments and Objective Measures of Lumbar Spine
Mobility Before and after Treatment”, Journal of Neurology, Orthopedic
Medicine, and Surgery, 1989, 10:141-144
3) Klein, Robert G., Mooney, Vert, ET AL “A Randomized Double-Blind Study
of Dextrose-Glycerin-Phenol Injections for Chronic Lower Back Pain”,
Journal of Spinal Disorders, 1993, 6:23-33.
4) Lui, Y Kinget Al., “An in-Situ Study of The Influence of Sclerosing Solution
in Rabbit Medical Collateral Ligaments and it’s Injection Strength”,
Connective Tissue Research, 11 (1983), PP 95-102
5) Ongley, Milne J., “A New Approach to the Treatment of Chronic Low Back
Pain”, The Lancet (July 18, 1987), PP 143-146
6) Dorman, Thomas A, Editor, Prolotherapy in the Lumbar Spine and Pelvis
Spine: State of Art Reviews, Volume 9 Number 2, Philadelphia, Hanley and
Belfus, Inc., 1995
7)Hackett, George S., and Hemwall, Gustav A., and Montgomery, Gerald A.,
“Ligament and Tendon Relaxation Treated by Prolotherapy”, 5th Edition, 2nd
printing, Oak Park, Illinois 1993
8)Houser, Ross, “Prolo Your Pain Away”, Curing Chronic Pain with
Prolotherapy”, Beulah Land Pres, Oak Park Illinois 1998.
9)Reeves, K Dean, “Technique of Prolotherapy”, Psychiatric Procedures,
Lennard, Ted A., Editor, PP 57-70, Philadelphia, Hanley Belfus, 1995
10)Wilkinson, Harold A., “The Failed Back Syndrome: Etiology and Therapy”,
New York, Springer-Verlag, 1992, PP 147-169.
Patient Preparation for Injections: 1)Read the Prolotherapy handout and make sure you have ALL your questions
answered before beginning treatment.
2)You may have a light meal the day of your treatment, however, please do not
consume any food within 2 hours of your treatment. The weeks leading up to
treatment you want to eat a lot of foods rich protein and vitamin C; this will
help with healing. Lots of high quality grass fed beef, pasture raised chicken,
wild caught fish, organic vegetables and fruit. You also want to make sure you
stay adequately hydrated; at least 64 fluid oz water/day.
3)For your safety it is highly recommended to have someone drive you home
after your treatment, mandatory if you are having treatment on your Neck.
4)Please STOP Aspirin and Non-Steroidal Anti-Inflammatory Medications.
It is Okay to continue taking “BABY-ASPIRIN” (81MG) unless otherwise
told by your doctor.
5)STOP ANTI-PLATELET AGENTS (PLAVIX, TICLID) for 7 days prior.
With approval from your doctor.
6)STOP COUMADIN (WARFIN) for 5 days prior to your injection with
PRIOR APPROVAL from your DOCTOR and obtain STAT BLOOD
TEST (PT, INR AND PTT) on the morning of your injection. Have any
results faxed to our office.
Please contact us prior to your appointment if:
1)You have a significant change in your medical history. 2)You have any questions about your medications. 3)You are taking antibiotics, are being for an infection or are feeling ill.
Thank you and we look forward to seeing you at your next visit.
Am I a candidate for Regenerative Injection Therapy (Prolotherapy)?
Many problems related to musculoskeletal conditions are can be successfully
treated with prolotherapy:
1.Degenerative Joint Diseases (Osteoarthritis) of all locations:
neck, TMJ, shoulders, scapulaes, clavicles, ribs, elbows, wrists, fingers,
thoracic and lower back pain, “disc’s problems”, hips, knees, ankles, feet,
toes.
2.Consequences of ligamentous and tendinous injuries (with some
limitations). Partial tears of tendons, muscles and ligaments can be
successfully treated with prolotherapy +/- PRP (platelet rich plasma)
3. Some forms of arthritis may have infectious, allergic, autoimmune or
metabolic causes. In such cases we will order special diagnostic tests to help
patient in selection of appropriate treatment.
4. Patients with infected joints, non-healing wounds around joints, cancer
spread into the bones (metastasis, multiple myeloma, leukemias) are not
candidate for prolotherapy
5. Elderly and debilitated patients may not respond well to prolotherapy.
6. Degree of joint damage (stage of the OA), age, weight, smoking status,
nutritional status, use of certain medication (Steroids , NSAID) will
negatively affect prognosis of the treatment.
7. Other factors may play role too: anticoagulation therapy, anti-platelets
therapy, etc
How much does it cost?
A.: The initial evaluation cost is $375.00. Treatment prices vary by area.
Detailed pricing will be given at consultation
Will my insurance pay for this treatment?
A.: No, unfortunately prolotherapy is not covered by insurance
How many treatments will I need?
A.: It is not possible to tell ahead of time how many treatments a patient
might need before they are pain-free. The doctor will give an estimated range
of the number of treatments that you will need, depending on the severity of
your condition. Average 3-6 sessions.
How far apart are the treatments?
A.: Treatments are usually given at 4-6 week intervals. There are exceptions
to this, depending on other circumstances.
How soon after treatment can I work out or play sports?
A.: If your sports or work-outs involve the area that is being treated, you will
get the best results with treatment when you avoid exercising or stressing the
area until 3 or 4 weeks after the last treatment. If you must continue to
exercise the treatment area, it may take a lot more treatments to get the
desired result. You may continue to re-injure it with the exercise or sports
activity, preventing it from getting strong enough to protect it.
What is in the medicine that is used in the injections?
A.: There is no cortisone used in prolotherapy. The solution is normally a
mixture of a very concentrated dextrose (glucose) with a local anesthetic like
lidocaine. In some cases we do PRP (Platelet Rich Plasma) which is your
own blood product.
What is the success rate with prolotherapy?
A.: Prolotherapy generally has about a 80% good to excellent response
among the doctors across the country that keep track of their patients’
responses to treatment. About 10% of the patients are in the poor response, or
less than 50% improvement category.
Is there a guarantee that prolotherapy will work for me?
A.: There is nothing in medicine that is guaranteed.
How do I contact the office for an appointment?
A.: Please call our office at 508-754-9950 for an appointment. The staff will
be happy to schedule your appointment and give you further information.
What are the office hours?
A.: Our office hours are 8:00 AM – to 5:00 PM, Monday through Friday.
What should I bring to the doctor’s office for the initial evaluation?
A.: Please bring the completed paper work that the office sends to you and
any X-ray or films of any other studies that you may have had.
What happens at the first visit?
A.: Medical assistant will greet you, collect your paper work and take you to
the examination room. The doctor will review your paper work, ask you
questions about your problem, examine you, read your X-rays and explain
them to you, give you their opinion about what they believe is causing your
pain or other problems, order any new studies that may be needed and make
recommendations for treatment. They will explain the treatment and answer
your questions. You will be given Consent form for treatment and estimated
cost. No treatment will be performed during consultative visit.
Will I need a driver?
A.: Most of the time patients do need a driver. You will receive a treatment
which may affect your ability to drive safely for 12-24 hours.
Will I need to be off work after the treatment?
A.: Most patients do not need to be off work the day after the treatment if it is
sedentary work.
What are the risks with prolotherapy?
A.: There are risks with all treatments and medications, if the doctor feels
that you are a candidate for prolotherapy, they will explain the risks to you
and try to answer all of your questions.
It is important to remember that prolotherapy is a controlled injury. Therefore
you should expect to be sore for the following 24-48 hours.
Orange Peel Tea for Immune Support During these uncertain times, supporting your immune system and the microbiome is important. You can drink this tea throughout the day and swish it around your mouth (unsweetened) to support the oral and gut microbiome. Ingredients: Orange or any citrus peel (from 4-5 fruits)1/4 Onion1 inch Ginger2-3 Sprigs of Rosemary2 tsp Whole Cloves2-3 Star Anise1-2 tbs Fennel seeds 8-10 cups water. Bring to a boil & simmer for 20 minutes (or up to an hour) Keep in mind that you don’t have to have every single ingredient. There is no way to do this wrong. A simple tea of citrus peel is helpful. The other ingredients can be rotated. The goal is to use what you have on hand and get lots of polyphenols from these plants. Boiling these plants in water helps get the nutrients out. You can use a vegetable peeler to peel the citrus and save the peels in a bowl as you go in your fridge. Once you have enough, you can make tea. If you are having digestive issues, prioritize the ginger and fennel after the citrus peel. The onion provides quercetin, a flavonoid, useful for immune support, and is very antimicrobial. You don’t taste it in the tea, but it provides benefits. The spices all function to support the gut flora. Star anise contains shikimic acid and is used as a base material for the production of Tamiflu.
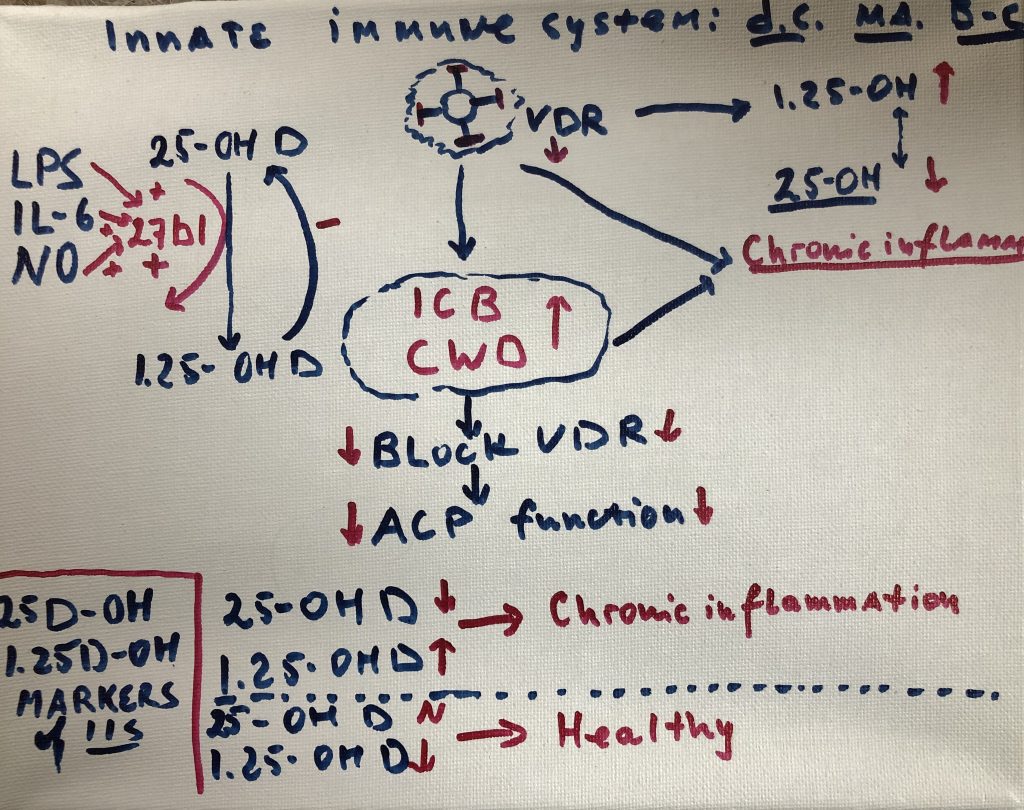
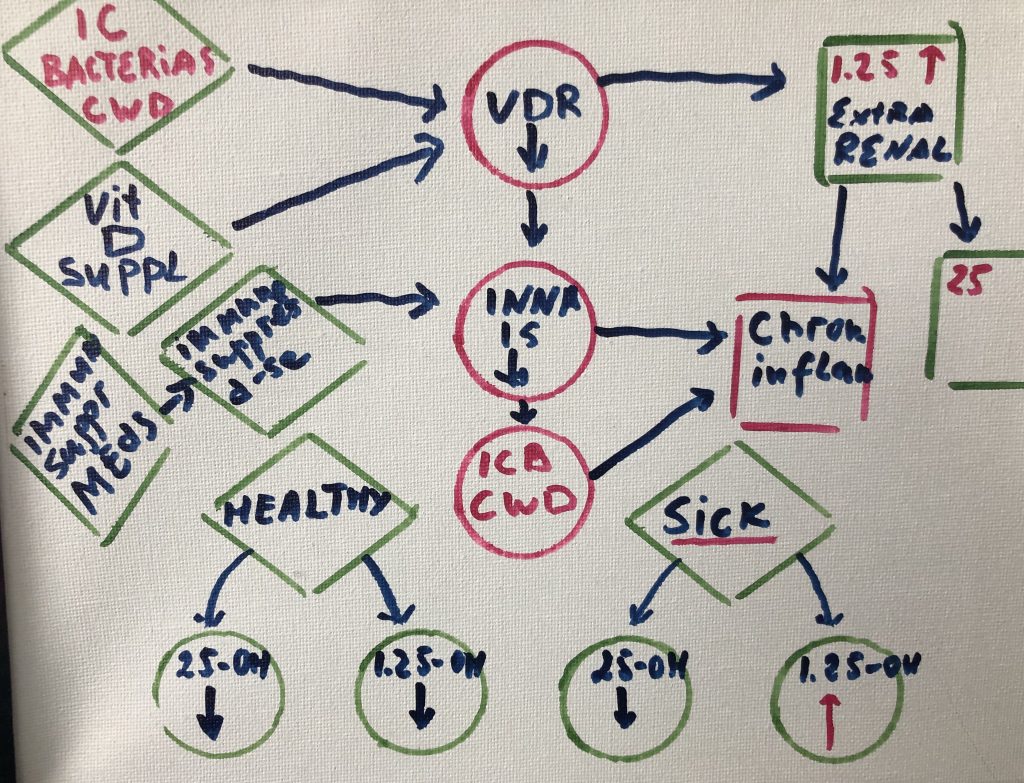
Cellular components of the organs and tissues, vascular and neurological structures are parts of tensegrity arrangement; Microbiome is an equal part of this ecosystem. Tensegrity is influenced by the microbiome. At the same time, mechanical properties of the connective tissue will influence microbiome composition and pathogenicity.
Cellular components of the organs and tissues, vascular and neurological structures are suspended in the tensegrity network and actually part of this network. Their form and function regulated by mechanical properties of the ecosystem they reside (Pre-stress).
Pre-stress is influenced by
microbiome and microbiome is regulated by pre-stress.
Microbiota are
ecological communities of commensal,
found in and on all multicellular organisms.
Microbiota includes bacteria, archaea, protists, fungi
and viruses. Microbiota have a crucial role
for immunologic, hormonal and metabolic
homeostasis of
their host. The synonymous term microbiome describes
either the collective genomes of
the microorganisms that reside in an environmental niche or the microorganisms
themselves
Microbiome
persist in nearly every human body site, including tissue and blood. The
genomes of these microbes continually interact with the human genome in order
to regulate host metabolism.
Many components of this microbiome are capable of both
commensal and pathogenic activity. This activity is determined by environment they
reside (viscoelasticity -pre-stress): immunological cells responsible for
innate and adaptive responses. They are additionally able to persist in both
“acute” and chronic forms. Inflammatory conditions historically
studied separately (autoimmune, neurological and malignant) are now repeatedly
tied to a common trend: imbalance or dysbiosis of these microbial ecosystems.
Collective activity of the
microbiome that drives inflammatory processes via complex microbe-microbe and
host-microbe interactions. Many microbes survive as polymicrobial entities in
order to evade the immune response. Pathogens in these communities alter their
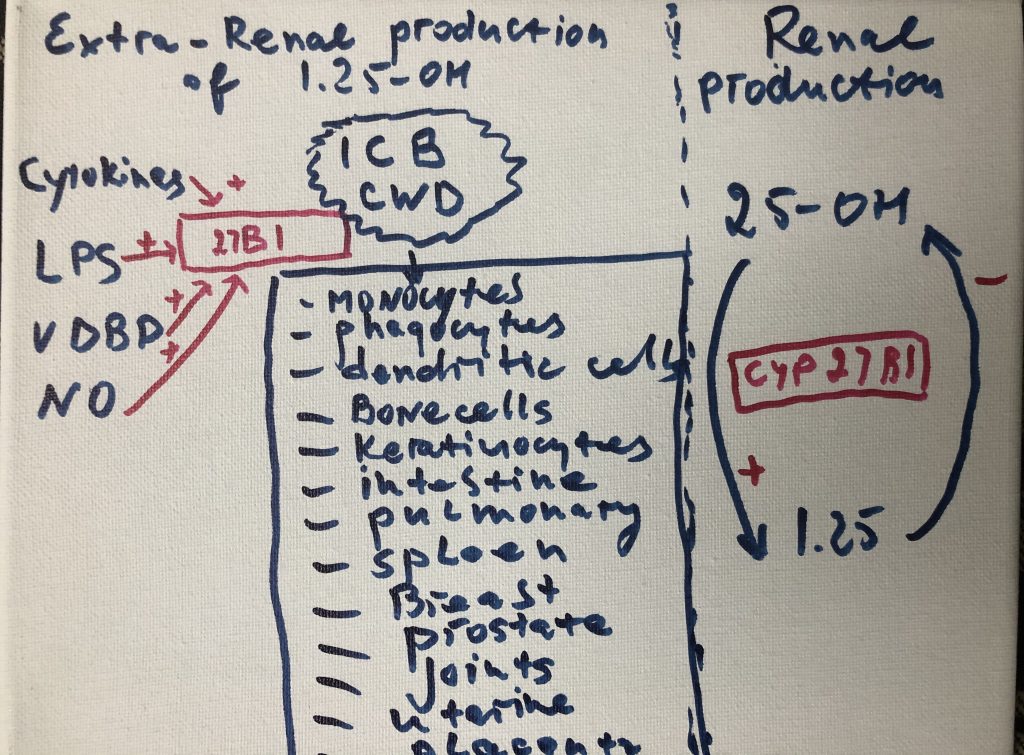
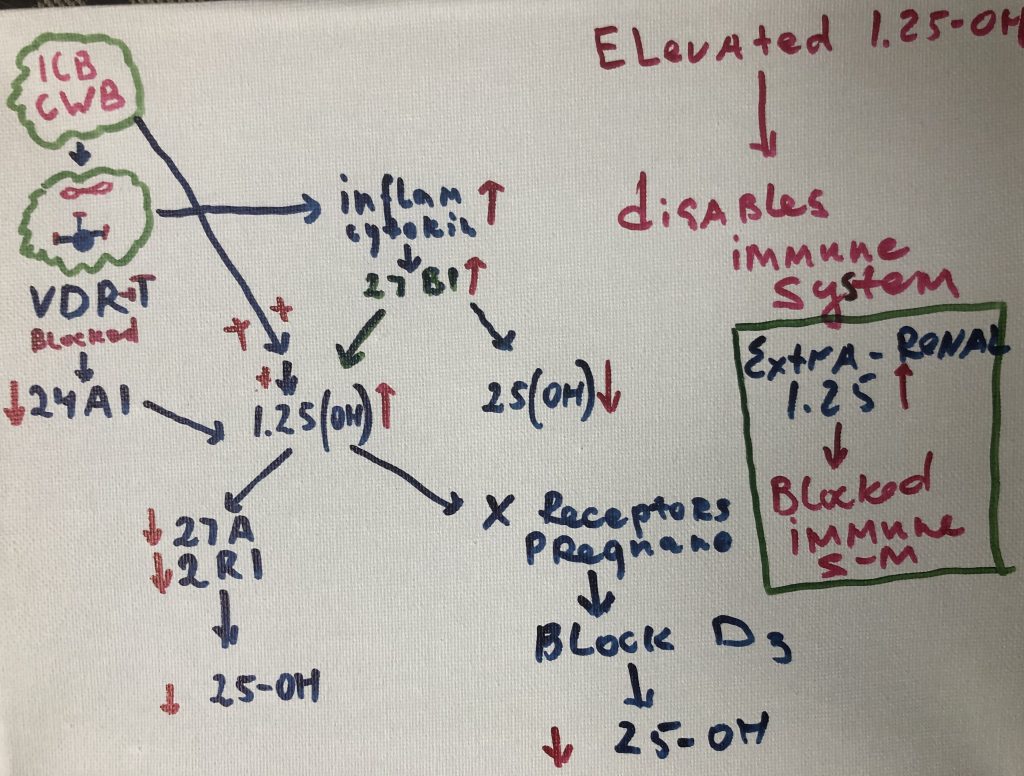
gene expression in ways that promote community-wide virulence. Other microbes
persist inside the cells of the immune system (Cell Wall Deficient bacteria -CWD),
where they directly interfere with host transcription, translation, and DNA
repair mechanisms. The numerous proteins and metabolites expressed by these
pathogens further dysregulate human gene expression in a manner that promotes
imbalance and immunosuppression. Molecular mimicry, or homology between host
and microbial proteins, complicates the nature of this interference. When taken
together, these microbe-microbe and host-microbe interactions are capable of
driving the large-scale failure of human metabolism characteristic of many
different inflammatory conditions.
Microbiota are “ecological communities
of commensal, symbiotic and pathogenicmicroorganisms” found
in and on all multicellular organisms. Microbiota includes bacteria, archaea, protists, fungi
and viruses. Microbiota have been found to be crucial for immunologic, hormonal
and metabolic homeostasis of
their host. The synonymous term microbiome describes
either the collective genomes of
the microorganisms that reside in an environmental niche or the microorganisms
themselves
Microbiome
persist in nearly every human body site, including tissue and blood. The
genomes of these microbes continually interact with the human genome in order
to regulate host metabolism.
Many components of this microbiome are capable of both
commensal and pathogenic activity. They are additionally able to persist in
both “acute” and chronic forms. Inflammatory conditions historically
studied separately (autoimmune, neurological and malignant) are now repeatedly
tied to a common trend: imbalance or dysbiosis of these microbial ecosystems.The collective activity of the microbiome that drives inflammatory processes via complex microbe-microbe and host-microbe interactions. Many microbes survive as polymicrobial entities in order to evade the immune response. Pathogens in these communities alter their gene expression in ways that promote community-wide virulence. Other microbes persist inside the cells of the immune system, where they directly interfere with host transcription, translation, and DNA repair mechanisms. The numerous proteins and metabolites expressed by these pathogens further dysregulate human gene expression in a manner that promotes imbalance and immunosuppression. Molecular mimicry, or homology between host and microbial proteins, complicates the nature of this interference. When taken together, these microbe-microbe and host-microbe interactions are capable of driving the large-scale failure of human metabolism characteristic of many different inflammatory conditions. (Amy D Proal)
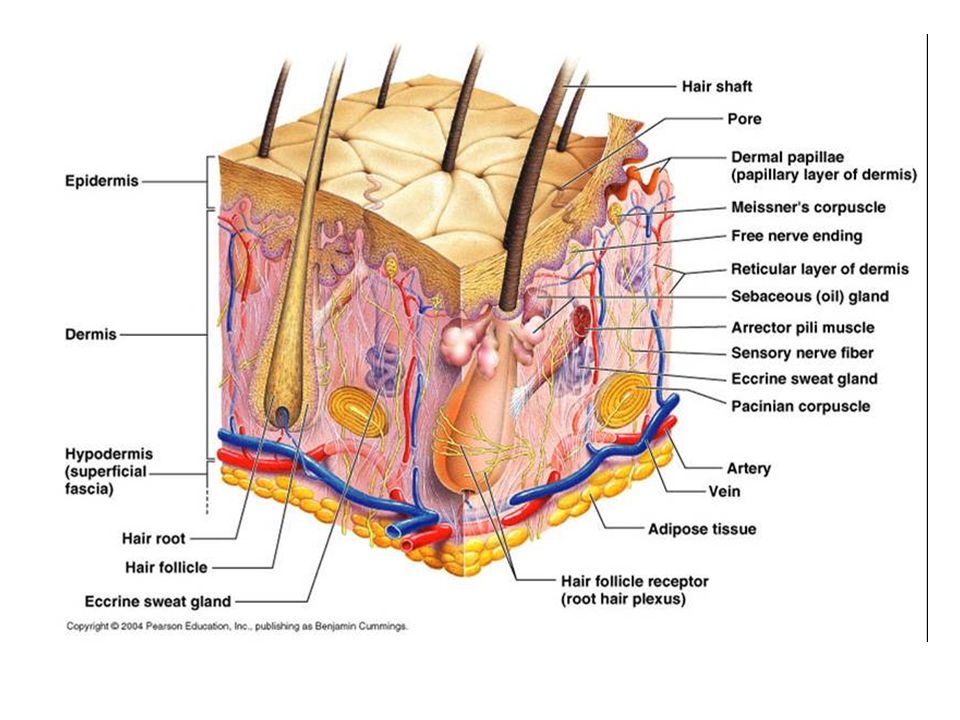
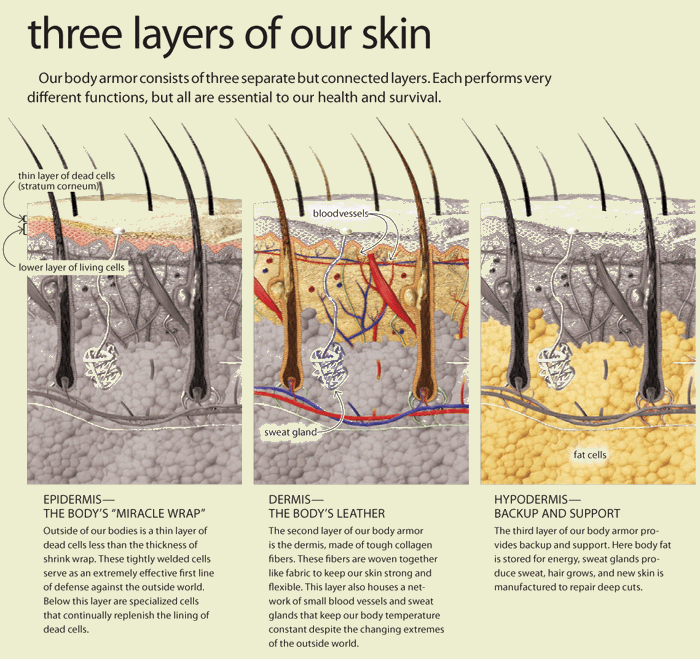
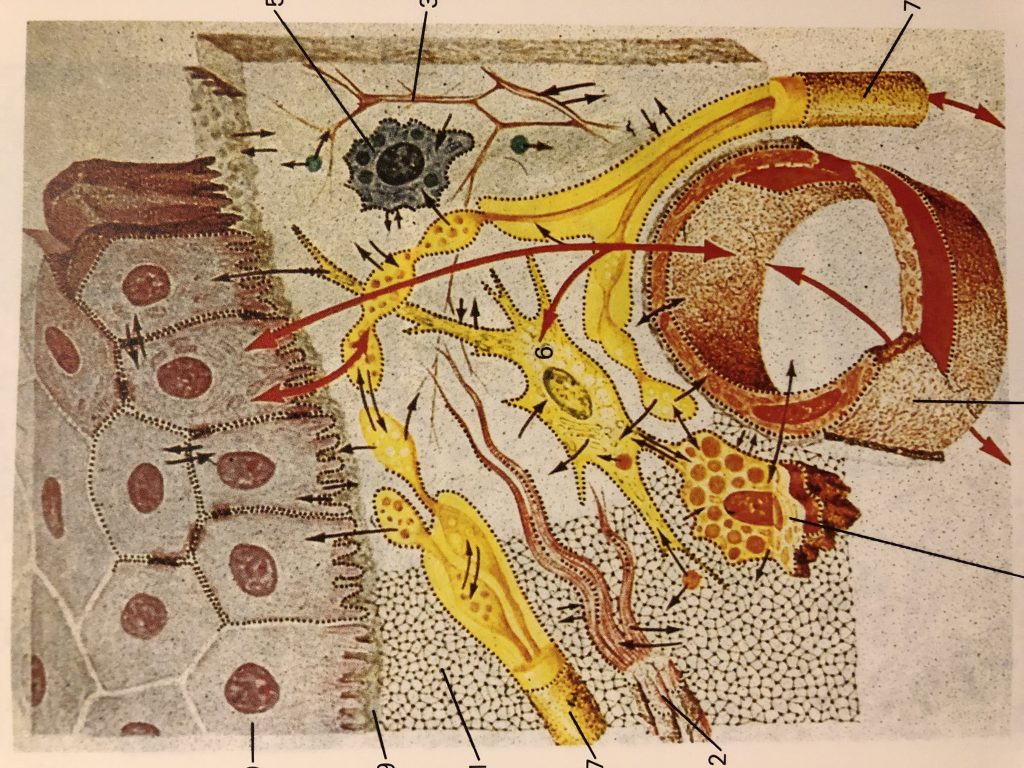
Mechanical properties of the skin are changed with time. Altered pre-stress changes first defenses of the immune system-physical barriers-Epidermis and dermal layers of the skin. Reduced functional capacity and increased susceptibility of the skin with development of dermatoses such as dry skin, itching, ulcers, dis-pigmentation, wrinkles, fungal infections, as well as benign and malignant tumors are the most common skin conditions in aged population. In turn altered appearance, dry skin, chronic wounds, and other conditions decrease general health and reduce the likelihood for healthy and active aging. Ability of the skin to carry out multiple and wide-ranging roles is very closely related to its structure, which is composed of an outer epidermis overlying an inner dermis, separated by a basement membrane.
Biochemical barriers: acidic, hydrolipidic nature of the
skin, as a result of sweat, sebum, lipids, and antimicrobial peptides
(AMPs).Any changes in lipid composition and epidermal differentiation lead to a
disturbed skin barrier, which plays a role in the pathogenesis of several
immune-mediated skin pathologies, such
as atopic dermatitis and ichthyosis vulgaris .
The epidermis is a host to keratinocytes, melanocytes
and immune cells such as Langerhans cells (LCs) and T lymphocytes, nerve-ending
cells (Merkel cells).The dermis is composed of an upper papillary (stratum
papillare) and lower reticular (stratum reticulare) dermis containing thin and
thick collagen fibers, respectively. The collagen fibers offer a mechanical
barrier as well as a structural environment
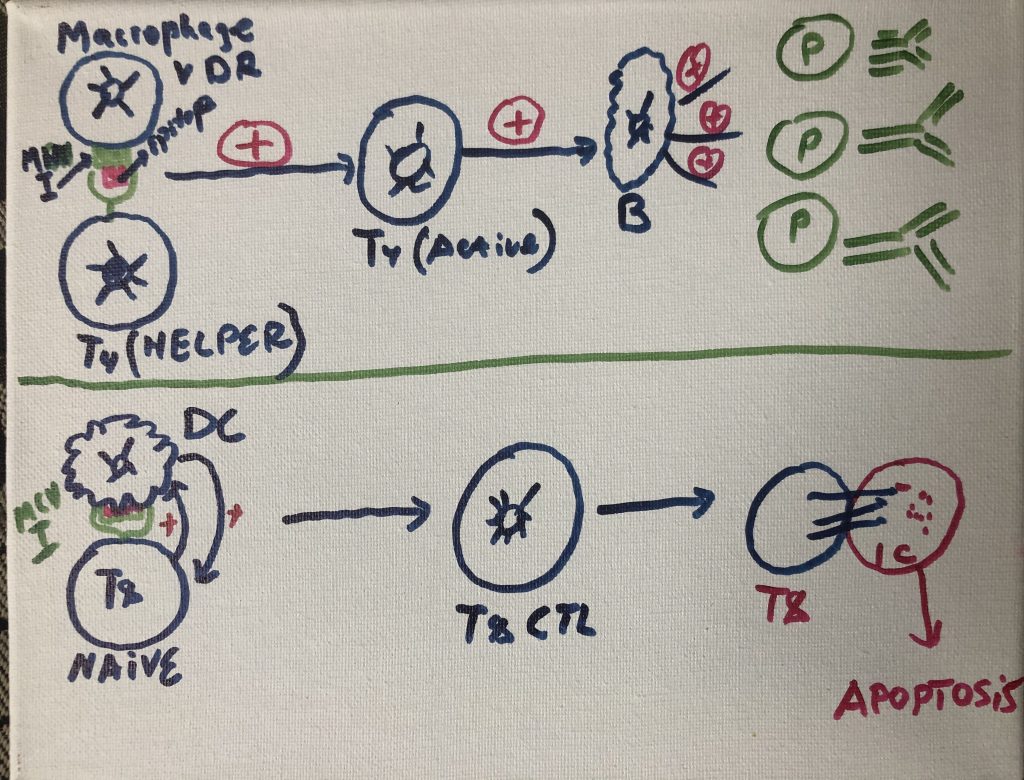
in which to host blood vessels and many immune cells such as dermal
dendritic cells (DDCs), ab T cells, gd T cells, natural killer (NK) cells, B
cells, mast cells, and macrophages can perform their function.
More recently, it was proposed a sentinel role in health
and disease for a spectrum of skin-resident cells ( with keratinocytes involved
in sensing pathogens and danger signals, migratory DCs capable of initiating a
diverse range of immune responses, and tissue-resident memory T (Trm) cells
performing crucial effector functions.
Skin-associated lymphoid tissue
(SALT) participates in trafficking of
immune cells between the skin, draining lymph nodes (LNs), and the peripheral
circulation (Streilein, 1983).
There is currently no vaccine to prevent coronavirus disease 2019 (COVID-19).
The best way to prevent illness is to avoid being exposed to this virus.
The virus is thought to spread mainly from person-to-person.
Between people who are in close contact with one another (within about 6 feet).
Through respiratory droplets produced when an infected person coughs or sneezes.
These droplets can land in the mouths or noses of people who are nearby or possibly be inhaled into the lungs.
Take steps to protect yourself
Clean your hands often
Wash your hands often with soap and water for at least 20 seconds especially after you have been in a public place, or after blowing your nose, coughing, or sneezing.
If soap and water are not readily available, use a hand sanitizer that contains at least 60% alcohol. Cover all surfaces of your hands and rub them together until they feel dry.
Avoid touching your eyes, nose, and mouth with unwashed hands.
Cover your mouth and nose with a tissue when you cough or sneeze or use the inside of your elbow.
Throw used tissues in the trash.
Immediately wash your hands with soap and water for at least 20 seconds. If soap and water are not readily available, clean your hands with a hand sanitizer that contains at least 60% alcohol.
Wear a facemask if you are sick
If you are sick: You should wear a facemask when you are around other people (e.g., sharing a room or vehicle) and before you enter a healthcare provider’s office. If you are not able to wear a facemask (for example, because it causes trouble breathing), then you should do your best to cover your coughs and sneezes, and people who are caring for you should wear a facemask if they enter your room. Learn what to do if you are sick.
If you are NOT sick: You do not need to wear a facemask unless you are caring for someone who is sick (and they are not able to wear a facemask). Facemasks may be in short supply and they should be saved for caregivers.
Clean and disinfect
Clean AND disinfect frequently touched surfaces daily. This includes tables, doorknobs, light switches, countertops, handles, desks, phones, keyboards, toilets, faucets, and sinks.
If surfaces are dirty, clean them: Use detergent or soap and water prior to disinfection.
To disinfect:
Most common EPA-registered household disinfectants will work. Use disinfectants appropriate for the surface.
Options include:
Diluting your household bleach.
To make a bleach solution, mix:
5 tablespoons (1/3rd cup) bleach per gallon of water
OR
4 teaspoons bleach per quart of water
Follow manufacturer’s instructions for application and proper ventilation. Check to ensure the product is not past its expiration date. Never mix household bleach with ammonia or any other cleanser. Unexpired household bleach will be effective against coronaviruses when properly diluted.
Alcohol solutions.
Ensure solution has at least 70% alcohol.
Other common EPA-registered household disinfectants.
Products with EPA-approved emerging viral pathogens pdf icon
[7 pages]external icon claims are expected to be effective against COVID-19 based on data for harder to kill viruses. Follow the manufacturer’s instructions for all cleaning and disinfection products (e.g., concentration, application method and contact time, etc.).
Spirulina is a blue green microalgae that grows in waters of subtropical climates.It is a type of cyanobacteria (cyano – blue pigment) that can be consumed as a superfood. Spirulina is has countless uses as a supplement for maintaining good health and preventing diseases. This is because it contains a plentiful supply of many important nutrients and antioxidants, including protein, complex carbohydrates, iron, and vitamins A and K, as well as B complex. It’s also rich in chlorophyll, fatty and nucleic acids, and lipids. Spirulina is rich in gamma-linoleic acid, or GLA, a compound found in breast milk that helps develop healthier babies. In fact it is often referred to as “Nature’s Multivitamin”.This is a breakdown of some of spirulina’s most significant benefits and nutrient composition.
Beta-Carotene
Spirulina contains powerful carotenoids such as best-carotene and yellow xanthophyll. It is one of the richest beta-carotene foods. The beta-carotene found in spirulina is ten times more concentrated than in carrots.
Iron
Spirulina is rich in iron, magnesium and trace minerals and is easier to absorb than iron supplements. And without the common side effects of iron supplementation.Ten grams of spirulina can supply up to 70% of the minimum daily requirements for iron.
Protein
About 60% of Spirulina’s dry weight is protein, which in the form it comes in within spirulina, is essential for growth and cell regeneration.
Gut Flora
Spirulina suppresses bad bacteria like E. coli and stimulates beneficial flora like lactobacillus and bifidobactria in the digestive tract to promote healthy digestion and proper bowel function.
Healthy flora is one of the foundations of good health. It increases the body’s ability to absorb nutrients from the foods we eat and helps protect against infection.
Detoxifier
Spirulina has a completely unique composition of phytonutrients, including chlorophyll, phycocyanin and polysaccharides, that can help to purge toxins from the body. In 1994, a Russian Patent was awarded for spirulina, deeming it a medical food for reducing allergic reactions from radiation sickness. This was a result of 270 children in Chernobyl consuming five grams a day for 45 days. Radionuclides were lowered by 50% and allergic sensitivities were normalized.
Cardiometabolic Benefits
Spirulina has been demonstrated to have cardiometabolic benefits – improving glycemic, lipid and blood pressure parameters.In a double-blind placebo controlled trial, overweight patients with hypertension were randomly allocated to receive 2g of Hawaiian spirulina daily for 3 months.The subjects that received spirulina exhibited a reduction in systolic blood pressure and some reduction in BMI (Body Mass Index).
Lipids
Spirulina also has a favorable effect on cholesterol.
A meta-analysis also showed that spirulina reduced plasma concentrations of total cholesterol, LDL and triglycerides.
Skin
Spirulina contains Vitamin E, selenium and tyrosine, which are all known for their powerful anti-aging effects.The antioxidants present in spirulina can aid in skin healing and support.
Quick Tip:
Adding 1-2 teaspoons of spirulina to your daily diet can be very beneficial.Try adding some to your smoothie or simply sprinkle a teaspoon into coconut water.
The flu, or influenza, is a viral illness that affects the respiratory tract including your nose, throat, lungs and bronchi. Mild cases of the flu can be confused with the common cold, however, the flu usually causes a more serious illness. Although it’s tempting to rely solely on natural remedies, the flu can be very serious, and one should always talk with a qualified healthcare provider if you think you may have the flu. Completely avoiding getting sick during the flu season ay be unrealistic, but there are ways to support your body naturally.The goal is to have a recovery plan and good sign of health is the recovery process itself.
Healing Herbs & Supplements
There are a variety of powerful supplements you can use as weapons against a flu virus.
Olive leaf is great for anyone who is starting to feel sick and noticing flu-like symptoms.
Echinacea and goldenseal are two supplements to incorporate when you are fighting a flu. Echinacea has been used for immune system support for hundreds of years.
Oregano is a great herb for flu viruses.Oregano oil is very powerful and should only be used short-term.
Elderberry syrup is another incredible flu-fighting supplement.
Elderberry (Sambucus nigra) is a herb that has a long history of use as a folk remedy for colds, sinus infections, and the flu. In preliminary lab studies, elderberry extracts have been found to fight off viruses. Researchers believe that anthocyanins, compounds found naturally in elderberries, may be the active component that strengthens the immune system and blocks the flu virus from sticking to our cells.
Propolis can be a powerful immune system support and can be great supplements to take if you start to feel the beginnings of a flu arise.
All different kinds of teas can provide benefits for someone suffering with flu symptoms. Steeping a strong cup of rose hips tea and adding in ample lemon juice can be particularly powerful and incredible for sore throats.Rosehips provide a great source of bioavailable Vitamin C. A simple lemon, honey & ginger tea provides soothing minerals to support you while you are healing.
Lots of rest & sleep.Sleep can help us immensely, it is very important for healing.
Hydration & lots of fluids. This is critical. Hydration allows the body to flush viral toxins out of the body and to keep the mucus thinned out. Staying away from heavy cooked foods is also helpful so that you don’t use up too much energy on digestion, which can instead be focused on healing.
Healing Foods & Recipes
Healing Broth
This healing broth is great while fighting off a flu. This healing broth can be nourishing, supportive, and help keep you from picking up any unwanted virus. Thus its useful to have all winter.
4 carrots, chopped or 1 sweet potato, cubed
2 stalks of celery, roughly chopped
2 onions, sliced
1 cup parsley, finely chopped
1 cup of shiitake mushrooms, fresh or dried (optional)
2 tomatoes, chopped (optional)
1 bulb of garlic (about 6-8 cloves), minced
1 inch of fresh ginger root
1 inch of fresh turmeric root
8 cups of water
Optional: Chili peppers or red pepper flakes
Place all the ingredients in a pot and bring to a boil. Turn heat down to low and allow to simmer for about an hour. Strain for a light soothing drink or leave the veggies in to enjoy as a light healing soup.
Congee
This simple rice soup is easily digested and assimilated, it harmonizes the digestion and is extremely nourishing.
1 cup rice, 1 garlic clove, 3 inch piece of ginger, 6 cups water.
Combine the rice, ginger, garlic and water in a large saucepan. Stir well. Cover and cook on low heat for approximately 1.5 hours until the rice is very soft and the congee has thickened.
Turmeric Ginger Garlic Orange Juice Shot
This is a powerful combination of flu fighting ingredients
2 oranges, 2 garlic cloves, 2 inch piece of turmeric, 2 inch piece of ginger
Run the ingredients through a juicer.Drink like a shot.This is a very powerful preparation.
Herbal supplements have been studied as supportive treatments for type 2 diabetes.
Type 2 diabetes affects more than 20 million Americans.It is a growing epidemic that is now very common and even seen in children. When someone has type 2 diabetes, it needs to be controlled through managing blood sugar levels. Diet and exercise are most important.
Herbal Supplements for Type 2 Diabetes
Several herbal supplements show promise in supporting type 2 diabetes.
Curcumin
The compound curcumin, which is found in the spice turmeric, has been shown to both boost blood sugar control and help prevent the disease. In a nine-month study of 240 adults with pre-diabetes, those who took curcumin capsules completely avoided developing diabetes while a sixth of patients in the placebo group did.
Ginseng
Ginseng has been used as a traditional medicine for more than 2,000 years. Studies suggest that both Asian and American ginseng may help lower blood sugar in people with diabetes. One study found that extract from the ginseng berry was able to normalize blood sugar and improve insulin sensitivity in mice.
Fenugreek
Benefits of fenugreek for diabetes have been demonstrated in both animal and human trials. In one study of 25 people with type 2 diabetes, fenugreek was found to have a significant effect on controlling blood sugar.
Cinnamomum verum(true cinnamon)
Consuming about half a teaspoon of this variety of cinnamon daily can result in significant improvement in blood sugar, cholesterol, and triglyceride levels in people with type 2 diabetes.
Aloe Vera
This plant has been used for thousands of years for its healing properties. Some studies suggest that the juice from the aloe vera plant can help lower blood sugar in people with types 2 diabetes.Aloe is available in juice form and can also be purchased fresh.The fresh leaves can be peeled and added to smoothies.
Milk thistle
This flowering herb is found around the Mediterranean See.Its active component is called silybinin. Milk thistle may reduce insulin resistance in people with type 2 diabetes who also have liver disease.
Holy basil (Tulsi)
This herb is commonly used in India as a traditional medicine for diabetes. Studies in animals suggest that holy basil may increase the secretion of insulin. A controlled trial of holy basil in people with type 2 diabetes showed a positive effect on fasting blood sugar and on blood sugar following a meal.
Gymnema
Gymnema has been used in traditional medicine as a treatment for diabetes. The Hindi name for this herb translates to “destroyer of sugar.”
Modern pharmacological research seems to support this traditional wisdom. Compounds extracted from gymnema have been shown to reduce the absorption of sugar from the intestinal tract and boost insulin production, all of which could help lower blood sugar. Furthermore, an active component of gymnema called gymnemic acid binds to the taste receptors on your tongue that perceive sweetness. As a result, it makes sweet things taste a lot less sweet.Animal testing confirms that gymnema reduces blood glucose levels.
Healing Foods Changing the diet is vital to managing Type 2 Diabetes.Eliminating processed foods, oils and sugars is critical, not only for diabetics, but for everyone.
Wild blueberries, papayas, blackberries, and raspberries are top fruits to eat if you have type 2 diabetes. Vegetables to focus on include spinach, celery, sprouts, kale, and asparagus. These foods help detoxify the liver, strengthen glucose levels, support the pancreas, boost the adrenal glands, and stabilize insulin.